 |
 |
|||||||||||
|
|
|||||||||||
|
Information Theory and CreationismSpetner and Biological InformationBy Ian MusgravePosted August 17, 2003 IntroductionDr. Lee Spetner is an information theorist who has written a book claiming that random mutations can not produce the kind of "informational" changes in biology that is allegedly required by evolution (1). It is interesting that in a book supposedly about information theory, the classic formulation of information theory of Shannon and Weaver (2) does not get mentioned. Spetner's notion of information in biology has been taken up by several groups of evolution deniers, and while others have produced specific critiques of his work (3,4), there is no overall general analysis of his arguments. In this review I will consider if Spetner's metrics can be validly applied to biology, and how Spetner actually applies them to real world examples. Although his arguments are superficially plausible, a closer look with some knowledge of biochemistry shows significant flaws. I will first briefly describe Spetner's metric of information, I will then show that 1) Spetner's metrics depend on a binding mechanism that does not occur in nature, 2) that Spetner's metrics require that substances bind to enzymes in an all or nothing fashion, whereas real substrates do not bind in this way. Furthermore, I will show that Spetner himself is inconsistent in his application of his metrics. In his Xylitol example he does not actually use the measure he develops, and in the streptomycin example he swaps to a different metric, when his original metric would show increased information. Finally, I will show that his "directed evolution" model is based on a misunderstanding of one form of random mutation.
Glossary
Overview of Spetner's MetricsClassical information theories such as Shannon's Information theory has been applied to biology by several authors. Those that have addressed evolution have concluded that evolution can, under appropriate circumstances, increase "information" in Shannon's sense (eg see 13). Dr. Spetner however claims that random mutations cannot increase "information". In his claim, Spetner uses two separate metrics of information. One is an "expectation" measure whereby an ensemble of different strings has less information that an ensemble of identical strings. This will surprise people familiar with standard Shannon Weaver information or Algorithmic information, but is a valid formulation under particular circumstances. I will not go into this in any depth because Spetner himself doesn't. He also uses an "addressing" measure of information. This is fairly simple to
understand. There is a formal way to do this using binary addresses for matrix elements in n x n (or n x n x n) matrices. It's a bit of a stretch mapping this onto substrate binding, but there is nothing inherently more loony about this than mapping telegraphy transmission to DNA replication. A key feature of this measure is that "information" is directly related to string length. That is, a longer string has more information in both the "word enzyme" (see below) and binary addressing formulations. Another important feature is that binding is "all or nothing". I discuss the implications of this in detail in Spetner's word-enzyme example. Enzyme-substrate binding and Spetner's specificityEnzymes are protein catalysts that promote chemical reactions on the compounds that bind to them (their substrates). As enzymes bind strongly only to a very few chemicals out of the multitudes present in their environment, they are considered to be highly specific. Spetner claims that enzyme specificity is analogous to addressing specificity, and that enzymes with many substrates have less information than enzymes with one substrate. In his explanation uses a "word enzyme" thought experiment rather than a real example, and we will examine this argument in detail later. However, at first glance it seems like a reasonable argument. Enzyme-substrate (or hormone-receptor) binding is likened to a "lock and key" mechanism, where the substrate is the key and the enzyme is the lock. (important caveat, both the lock and key are "floppy" as enzymes and their substrates are somewhat flexible). 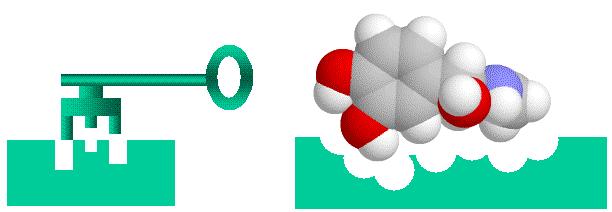
Figure 1: Binding of ligands to proteins like enzymes or hormone receptors has been likened to a "lock and key" arrangement, where the molecule fits into a space in the protein in a similar manner to a key fitting into a lock. While a useful analogy to help us visualise things, this image can be misleading as we will see, as various binding interactions are important, not just physical "lumps and bumps". Spetner's binding specificity is not only a property of enzymesNote that Spetner's argument is a general one, and applies not only to enzymes, but to any specific binding system, such as hormone/receptor interactions, binding between cytoskeletal elements etc, regardless of whether the substrate is acted on enzymaticaly. In the following discussion I will be using the term "ligand" which covers everything that binds specifically to an acceptor site, which includes substrates. Note further that there is a subtle shift in the argument. One can easily see that the address for "666, Richmond Road, Cannon Hill, Brisbane" takes more information to specify than "Brisbane", but the claim that enzymes have more information because they bind only one substrate is similar to the claim that "666, Richmond Road, Cannon Hill, Brisbane" has more information than "Brisbane" because fewer letters are delivered there than there are to "Brisbane". This is a somewhat different claim, that the information of the physical object at an address has the information required to specify that address. Spetner's requirements are not found in real world systemsBut accepting Spetner's claim, it might seem intuitively obvious that a more specific lock accepting only one key has more information a less specific lock which accepts many keys. This is actually incorrect, but rather than go into this here see this paper on cryptography and master keys: WARNING 5Mb file. For the moment we will accept this though. Analogously we might expect an enzyme is more specific because it has more binding points than an enzyme that is less specific (and there is also the unstated assumption that single substrate enzymes are more important than multi substrate enzymes. They are not). For example we might imagine an active site that uses 4 binding points has more information than one with two binding points X--O O--Y X--0 O--Y
\ / \ /
DRUG DRUG
/ \
Z--N N--Y
Where X, Y and Z are amino acids in the enzymes active site. Unfortunately, biology doesn't work that way. The number of binding points are important, to a degree. But other physical properties of the enzyme are important too. For example, take the betalactamase enzymes that break down the antibiotic cephalosporins. One mutant variant can breakdown extended cephalosporins (where the molecule has been made more bulky), where as the normal enzyme can't. The mutant variant doesn't have fewer binding points than the normal enzyme, but has a more flexible hinge so that the catalytic group can reach the lactam ring easier. Worse still, you can have substrates which bind at exactly the same number of points, but which bind more firmly because you have substituted a chlorine atom for an oxygen atom, and changed the distribution of electrons on the drug molecule. Binding specificity is not something simply analogous to "addressing", it involves physical shape (which is amenable to an addressing analysis) and physicochemical properties (which is not simply amenable to an addressing analysis, especially when the properties are those of the substrate) and structural properties of the enzyme, like hinge flexibility or helix distortion which are also not amenable to an addressing analysis (does a helix which flexes to the right have more information than a helix which flexes to the left?) Simply put, any analysis of the "information" of a protein that addresses the substrate binding specificity, although superficially plausible, is naive in the extreme, because substrate specificity is not amenable to the proposed analysis (especially when some of that information is in the substrate). Also, his analysis deals solely with single substrate enzymes, how he deals with bi-bi-random two substrate two product enzymes I don't know, he seems to feel that all enzymes should be like (some of) the enzymes of the Krebs citric acids cycle. Having outlined in general why Spetner's metric is not applicable to protein-ligand binding, in the next section I will examine Spetner's word enzyme example in depth, and compare this to a well studied receptor-ligand system, the angiotensin II receptor. Application to real biological systems:Spetner's word-enzyme exampleLets examine this in some detail and revisit his "word enzyme" example. This differs in key ways from the binary addressing example he gives first, but I'll ignore that for this article, as the "word enzyme" is the example he uses to bridge the gap between real systems [Streptomycin in his example] and the binary addressing measure, see (1) pages 134-137. Spetner's Information is proportional to binding string lengthHere is Spetner's word enzyme "ghts". This can "bind" many "substrates" (actually ligands, as this is a general binding argument): ghts Nights Lights Fights Rights (and many more, omitted for clarity) Lightship Nightshade by increasing the string length, we reduce the number of "ligands" "bound", increasing "specificity" ghtsh Lightship Nightshade by increasing the string length again, we reduce again the number of "ligands" "bound", increasing "specificity" yet again. ghtshi Lightship At each stage, for every "increase in specificity", the bit length of the binding string increases. Thus the "information" in the binding string increases. Note that although Spetner has claimed that random mutations cannot produce increases in information, in his "word enzyme" example random mutations can increase information. Simply randomly adding a letter to the string "ghts" will result in a number of strings that bind substantially fewer "ligands". It will also result in a number of stings that bind no ligands, but it is enough to note that simple random mutation will produced "word enzymes" of higher "specificity" However, when we come to real systems, the link between increased binding "string" length and "specificity" just doesn't hold. An example I have given above, a change in a hinge region, far from the binding site, can allow in bigger ligands, even though the actual binding sequence is unchanged. Skip text graphic a--d--------| a--d-------| a--d--------| abcd n VS abcd N abcdE N bc----------| bc---------| bc----------| In the section that follows I will examine this in greater depth. Spetner's Information depends on "all or nothing" bindingAs noted above, Spetner's information is proportional to number of substrates, but Spetner never defines substrate. This is not trivial. As we have seen above Spetner's "enzyme" either binds a ligand, or it doesn't. This is important as the whole nexus of Spetner's argument is that the number of substrates reflects the length of the binding string, and the length of the binding string, in bits, is the information of the enzyme/receptor/binding protein. However,in the real world the binding of a ligand to a protein is not an all or nothing affair, substrates have varying degrees of "stickyness", which is not addressed by Spetner's metric. Even a very specific enzyme will bind one ligand very tightly, a few not so tightly, and a great number very weakly. One cannot simply say that only very strongly bound ligands will be considered, as very weak ligands may be of great physiological importance, if they are present in a high enough concentration (there are many examples of this in physiology, in the angiotensin II receptor example below, there are a group of peptides called the histidine triad peptides, which though being very weak binders of the angiotensin II receptor, do indeed modulate the receptor activity under physiological circumstances as their concentrations are so high in the body). This has a theoretical and a practical implication. Theoretically, this means that there is no necessary connection between string length and the number of ligands bound, as "stickyness" depends on molecular factors that do not easily translate into information. Does a string containing aspartic acid have more or less information than one containing asparagine, when virtually the only difference between them is charge? "Stickyness" also depends on some molecular features that are part of the substrate. Practically, it means that Spetner information cannot be measured experimentally, since we can never truly know the total number of substrates unless we test all potential substrates in the universe. Thus we can see that Spetner's metrics have are unlikely to be able to measure the information content of enzymes/receptors/binding proteins at all. In the next section I make an in depth analysis that drives this point home. Word-enzymes compared to real proteinsLet's take as an example the important hormone receptors, the angiotensin II (AT) receptors. The AT receptors come in two versions, coded by separate genes, the AT1 receptor is protein made of a chains of a 359 amino acids, and the AT2 receptor is a 363 amino acid long protein. Both bind the peptide angiotensin II (AII) and related peptides, but AII binding to the receptors stimulates entirely different enzyme systems (AII binding to the AT1 receptor activates the enzyme phospholipase C and the binding to AT2 activates a different enzyme, phosphatase). Despite almost identical binding profiles, the receptors have only 34% of the amino acids in their structure in common. We have a very good understanding of the structure and properties of the AII binding site in the angiotensin receptors, so they provide a platform for testing Spetner's ideas about specificity. AII and similar ligands bind to the same amino-acid sequence in the AT1 and AT2 receptor. We can use the one letter amino acid code, where a single letter substitutes for an amino acid name, (eg D is the amino acid aspartate, see below), to represent the binding sequence as a string, analogous to Spetner's word binding string ghtshi. The angiotensin receptor binding string is DNKH. So these receptors look like a good example to test Spetner's ideas. The amino acids in the binding string are not contiguous, but are separated by many amino acids (5, 6). The amino acids that make up the sequence DNKH: D(Aspartate 281) N(Asparagine 111) K(Lysine 199) H(Histidine 256)
AT1 Note that the amino acid's are in different positions in the different receptors (eg , Aspartate 281 is at position 281 in the 359 amino-acid long chain that is the AT1 receptor and Aspartate 297 is at position 297 in the 363 amino-acid long chain that is the AT2 receptor) partly because the AT2 receptor has a longer C-terminus, and also due to an insertion into the sequence between N and K and between K and H in the AT2 receptor. Thus the concept of "precise sequence" is not exactly applicable to receptor binding. The amino acids are bought close (but not into a linear sequence, more like a ring) by the three dimensional folding of the proteins, and this three dimensional folding is bought about by very different sequences. 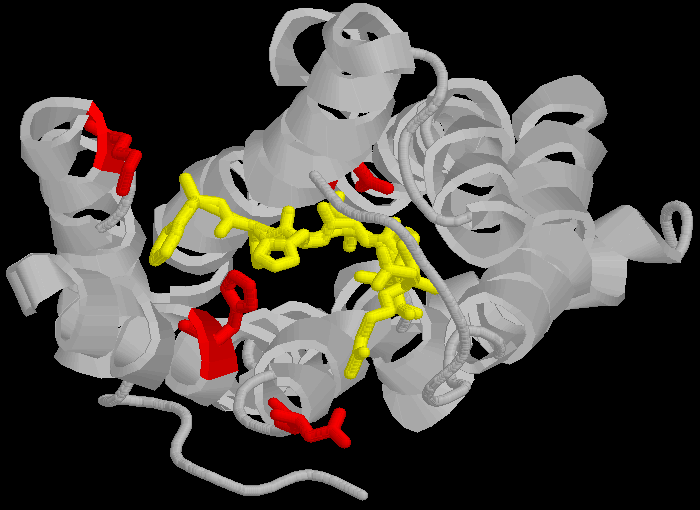
Figure 2: How proteins fold to give three dimensional structures that bring amino acids in proximity. Left hand panel, schematic showing how the helicies brings the DNKH sequence together (only 4 helicies shown for illustration purposes). Right hand panel, the three dimensional cartoon structure of the AT1-receptor with AII bound. We are looking at the receptor from above, with some chains removed for clarity. The transmembrane helicies are shown as grey coils, AII is shown in yellow, the DNKH amino acids are shown in red. See http://home.mira.net/~reynella/chime/ang_tuta.htm for 3D structures (needs MDL CHIME plug-in to view and manipulate the 3D structure). H H---H H--| ED EN EK EH | L L L L | I I I I | X X X---X | |--------------| This is problematic for Spetner's claims, as the sequence DNKH depends on a number of factors that are NOT dependent on that sequence, thus the "true" information content is not reflected by the binding sequence alone. Again note that the three dimensional folding is not rigid, and flips between a number of different states (Protein chains are somewhat flexible, more like a ball of semi-cooked spaghetti than the rigid shapes they are sometimes drawn as, thus enzymes and receptors are "floppy" locks and ligands are "floppy" keys: this is relevant to Spetner's claims, more later). As I said above, the AT receptors bind angiotensin and similar peptides to the ligand binding site. As with the receptor, not all of the ligand sequence binds to the binding site. The sequences are shown in the one letter amino acid code, with the amino acids that bind being shown in bold. AII DRVYIHPF AIII RVYIHPF SarIle SRVYIHPI SarAsp SDVYIHPI CGP NYKRHPI AIV VYIHPF For AII and AIII, the peptides bind to the receptor binding site sequence thusly (5,6,7,8): RYF Broadly analogous to ghtshi-lightship "binding". F actually binds to both K and H. This is mostly electrostatic interactions rather than the physical "bumps and hollows" Spetner uses in his (incorrect) analogy for streptomycin binding. The basic amino acid R binds electrostatically to the acidic amino acid D, Y and N form hydrogen bonds and F forms a pi bond with H, and a hydrogen bond with Y. (In SarAsp, the acidic D interacts with the acidic D, but dipole charge distribution means that they bind rather than repel. Chemistry and common sense do NOT go well together. (5,6) But as we look at SarIle and SarAsp, we can see Spetner's model of binding is going badly wrong. These have only two of the three matching points present in AII and AIII, yet they bind very well indeed. Maybe only the DN binding sequence is critical. AIV, which lacks the D binding region, still binds (not as well as the others, but it still binds, more on this later (5,6,7,8). In terms of Spetner's model, it is as if "ghtshi" bound Lights, Nights, and Ship as well as lightship. This breaks the nexus Spetner has tried to form between the binding string length and number of ligands bound. 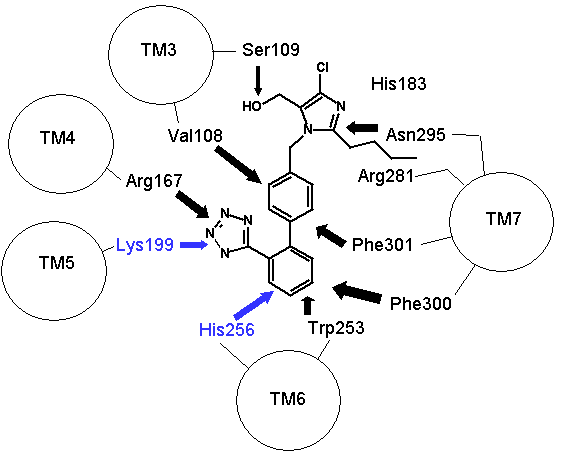
Figure 3: Binding of ligands to the AT1 receptor, looking down on the receptor from the top. Circles represent transmembrane helicies (see figure 2). Some transmembrane helicies have been omitted for clarity. Top panel, Binding of Angiotensin II to the AT1 receptor, the N terminal region of angiotensin is represented by R for simplicity. The D(Asp 281) N(Asn 111) K(Lys 199) H(His 256) binding sequence is shown in RED (as for figure 2), other amino acids that are peripherally related in stabilizing the molecule are shown in BLACK. Bottom panel: Binding of the ligand losartan to the AT1 receptor. The common binding sequence between AII and losartan is shown in BLUE, other amino acids that are peripherally related in stabilizing the molecule are shown in BLACK. Note that different amino acids are involved in losartan binding. Worse yet, CGP binds to completely different amino acid's in the ligand binding cleft, so there is a further disconnect from Spetner's model, as there is more than one "binding string" in a ligand binding site. This is not an unusual phenomenon, and is also true of many receptors. Figure 2 shows the comparison between the binding points of AII and losartan in the AT1 receptor, showing the differences. Numbers of ligands bound is not simply related to binding string lengthSo, what happens when we shorten the binding string DNKH. According to Spetner's model, you should increase the number of ligands bound. We can mutate the amino acid H to A (alanine), Q (glutamine) or R (arginine). The Q mutation keeps charge and size, but loses the pi bonding, the R mutation keeps the charge, the A mutation loses charge and size, thus mutations from H to R and A are like truncating the binding string to DNK. In the AT1 receptor, the binding doesn't change with any of these mutations. When we mutate H to Q or R in the AT2 receptor, all binding is lost (7,8). Either way, Spetner's model is dead wrong about what happens when the "string length" of a ligand binding string is shortened (and this again invalidates his measure). Even worse, what happens when you mutate N in DNKH (Asparagine at position 111 in the AT1 receptor and position 126 in the AT2 receptor)? If you mutate N to G (glycine) a very short neutral amino acid, unlike the longer charged asparagine, there is no contact with the ligand and the G, so the binding sitestring becomes DKH. Despite this truncation, you still bind AII, AIII, etc., but AIV has become "stickier"(something which Spetner doesn't actually address in his metric). As I mentioned before, the AT receptors flop between several conformational shapes. The N->G mutation restricts the receptor to fewer conformational shapes, making it MORE specific. It turns out that AII and AIII bind to all the conformational shapes that the enzyme flops between, but AIV binds only to one of the forms, that stabilised by the N111G mutation (5,6). Thus binding is a critical function of 3D shape, not covered by Spetner's metric, and a single mutation can increase specificity (as according to Spetner's criteria, a protein that has one conformational shape has more specificity than one with multiple conformational shapes). What if we take Spetner's metric at face value, looking ONLY at the numbers of ligands bound. Are there other examples of mutations that can increase "specificity" in the AT receptors. Yes. In the AT2 receptor, replacing the amino acid Y (tyrosine) at position 215 in the AT2 chain with R wipes out CGP binding, leaving the AII and SarIle binding intact (note that Y is NOT part of the DNKH binding string). Conversely, mutation of Y215 to Q wipes out AII and SarIle binding, while leaving the CGP binding intact (here "wipes out" means that there is no significant binding when 1 mM of liagand is present). Thus we can see that single mutations can increase "specificity", as in the number of ligands bound. Thus, even with Spetner's invalid metric, we can still show that random mutation increases "specificity". We have seen that in real world examples, Spetner's requirements for substrate number to be a indicator of protein information are not met, and Spetner's metric is invalid. We will now turn to the examples Spetner presented in his book, and show how his own analyses of these examples fail. Spetner's examplesLets now look at how Spetner himself applies his metric. Xylitol metabolismGiven the extensive discussion Spetner gives to binding specificity, and his thought experiments emphasising that specificity is equivalent to the number of substrates bound, it may come as some surprise that in Spetner's analysis of the ribulose to xylitol mutation he doesn't actually consider binding specificity. He does consider a biochemical measure called specificity, but this is not Spetner's specificity measure. This is a ratio between catalytic efficiency and binding specificity. Okay, you say now, this is pretty trivial, we can work with this. Well, no. Catalytic efficiency is something else again. It is definitely not amenable to an "addressing" measure, and is based on a range of physicochemical properties such as the charge distribution in the substrate, the free rotation of the catalytic side group (does a catalytic group that rotates 10 angstroms have more or less information than one that rotates 15 angstroms?) and so on. Critically, two substrates can have the same binding specificity, but different catalytic efficacy which confounds the information analysis. Worse again, Spetner's argument is related to the number of substrates acted on (see above). Yet in the ribulose dehydrogenase mutating to xylitol dehydrogenase, in both cases the enzyme bound 3 substrates, by his own definition no information change has occurred. The rate of dehydrogenation has changed, but the number of substrates acted on does not change. Two points need to be emphasised: a) As noted in the previous section, Spetner's metric is a measure that says an enzyme that binds substrate A and substrate B has less information than an enzyme that binds substrate A alone (remember ghtsh, which "binds" Lightship and Nightshade has less information than ghtshi which only binds Lightship, because ghtshi is a longer string). This does NOT work when we are comparing an enzyme that binds 20 molecules of substrate A for every 80 molecules of substrate B, with and enzyme that binds 80 molecules of substrate A for every 20 molecules of substrate B. The length of the binding string need not have changed, or changed in the direction that Spetner demands, so the bit length measure is no help at all. b) The relative magnitude of the change may be entirely due to changes in catalytic efficiency, which has no information content in Spetner's addressing scheme (nor can I see any meaningful way to calculate the "information" in this with any existing information metric). It gets worse still. Spetner compared the "specificities" of 3 substrates, because they were the only substrates measured in the experiment. In reality ribulose dehydrogenase (and xylitol dehydrogenase) binds a lot more substrates than just those 3 (although the catalytic efficiency is very low for the majority of substrates bound). As pointed out above, without assessing the full panel of substrates, any claim about specificity is meaningless. (But which substrates? if we restrict ourselves to natural substrates, we exclude xylitol, a synthetic sugar not found in the natural environment, but developing xylitol dehydrogenase activity was the whole point of the exercise) if we include synthetic substrates, we have a potentially infinite number of substrates to test. How do we know what is really happening? It continues to get worse. Spetner assumed that the point reached in the experiment (where Ribulose and Xylitol were being broken down at roughly similar rates by the mutant enzyme), was as good as it gets, no further improvement was possible. In the experiment he looked at, the main point was to see if xylitol activity could be developed, not getting the optimum activity. In fact, in other experiments mutant enzymes were produced that broke down xylitol 20 times faster than ribulose (which kind of destroys his thesis see 9). Thus the ribulose example does not support Spetner's thesis. Furthermore, we have literally hundreds of enzymes where random mutation results in high substrate specificity (there has been enormous amounts of work on developing novel and specific enzyme activities from generalist alpha-beta barrel proteins e.g. see 10, and recently the evolution of specific binding activities from random peptides has been reported 11). Streptomycin binding: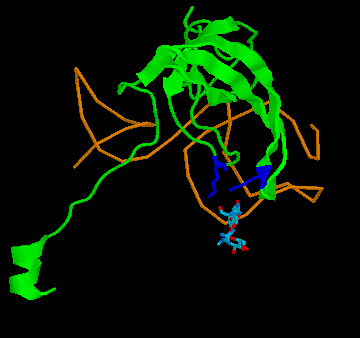 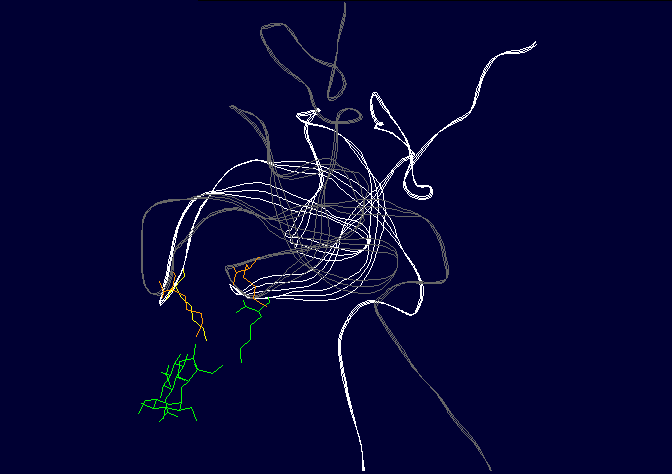
Figure 4: Binding of streptomycin to the S12 subunit of the 30S ribsomal subunit. Top panel, The S12 chain is shown in cartoon format (green), with the rRNA shown as the backbone only (brown). The residues critical for directly binding streptomycin, Lysine 42 and Lysine 87, are shown in blue. Streptomycin is shown with carbon as grey, nitrogen as blue and oxygen as red. Binding to the RNA is not shown for simplicity. Bottom panel: the effect of the mutation on S12 structure. The replacement of lysine 42 (yellow, on the white chain) causes the chain to twist away from the streptomycin (pink chain), preventing binding. His other example is the case of resistance to the antibiotic streptomycin. Streptomycin kills bacteria by interfering with protein assembly on the ribosome. Mutation of the rspL gene which codes for the S12 subunit of the 30S ribosomal particle in bacteria can result in resistance to streptomycin. The 30S ribosomal particle is a multi-subunit structure which in turn forms part of the protein synthesizing ribosomal particle. The S12 subunit together with the 16S RNA forms part of the proof reading center of the transfer RNA (tRNA) acceptor binding site. Mutation of streptomycin-binding lysine at position 42 in the protein chain that makes up the S12 subunit to threonine or asparagine results in streptomycin failing to bind to S12, with resulting resistance of the bacteria to the antibiotic streptomycin. This is a classic example of a beneficial mutation, as is found in many textbooks as it was work on this mutation that determined that mutations were random. Antibiotic-resistant mutant ribosomes have increased Spetner informationIn terms of Spetner's binding metric, the mutant S12 protein is more specific, it now only binds tRNA rather than tRNA and streptomycin. Now, streptomycin isn't a substrate, so you might object to using binding specificity in this case. However, Streptomycin binding is Spetner's own example for binding specificity, and he uses it as an example of a "lock and key" binding system (see above). Recall also that his metric is generally applicable to all ligand-acceptor interactions (remember that in his "word enzyme" example, there is just binding going on, so by his own examples the ligand doesn't have to be a substrate). Furthermore, the mutant does have an effect on the substrate-binding accuracy of the ribosome. While streptomycin binds at at different site from the actual tRNA proof reading site, this is a classic example of something called allosteric modulation. In Spetner's simple model of substrate binding, only ligands that directly bind to the active site are considered. However, many enzymes and hormone receptors have sites for ligands distinct from the active site, where binding of ligands to these distant sites results in modification of substrate/ligand binding at the active site. These kinds of ligands are called allosteric modulators, and are very important in biology. They include sodium binding to a pocket on protein-digesting enzyme trypsin, far from the active site, which alters trypsins catalytic activity and glycine binding to an modulatory site far from the glutamate binding site on the glutamate receptor (which alters nerve activity) and many other examples. If you exclude a lock and key binding allosteric modulator of enzyme action like streptomycin from consideration, then you are ignoring biology. Streptomycin-binding ribosomes turn out garbage proteins because streptomycin messes up the proof-reading centre (which is how streptomycin kills bacteria). The mutant version which doesn't bind streptomycin is actually MORE accurate, i.e. more SPECIFIC, than the wild type. The wild-type proof reading centre makes a few mistakes even in the absence of streptomycin, and the mutant forms make even fewer mistakes than the wild type( roughly 85% fewer; 12). This is a clear increase in Spetner's binding specificity:
Does Spetner acknowledge this? No, Spetner now swaps to the expectation measure and claims (without evidence) that since there must be more S12 sequences that don't bind streptomycin than those that do, information must have decreased (why didn't he do this analysis on the ribulose enzyme?). However, he is dead wrong. The ensemble of all rsPL genes that produce streptomycin binding S12 is around 1060. The ensemble of all rsPL genes that don't bind streptomycin is around 1060 too (from an analysis of neutral mutations using the method of Yockey (13). So the information difference is so small as to be non existent. If we only take amino acid changes in the streptomycin binding amino acids, then there are roughly the same number of substitutions that will allow binding as those that don't (10 resistant versus 6 normal see 14). Importantly, there is a particular mutation, AAA42 -> AGA42 (lysine to arginine) which doesn't bind streptomycin AND has wild type accuracy and translation rates(15). This is the only possible (single) mutation that does this, so it's ensemble is smaller than the wild type ensemble. Another example is the mutation that results in the S12 subunit still binding streptomycin and being resistant to streptomycin. Again, this is the only possible mutation, so by the expectation measure this mutation has more information than the wild type. Thus we see, that however Spetner applies his metrics, the Streptomycin-resistant mutation has more information that the wild type gene. Spetner and "directed" mutations:Spetner believes he has shown that random mutations can not produce the increases in information that would be needed in evolution (as we have seen, he is wrong). Spetner proposes that evolution is via "directed" mutations (with the implication that Divine Intelligence is in some way behind this "direction"). The apparent existence of "directed" mutations was based on some early work by Barry Hall. However, Spetner did not follow up the more recent research on this work, and misunderstands the origin and significance of "directed" mutations. Most people know that mutations arise at random in dividing cells, and that the mutations occur at various places in the replication cycle as a consequence of damage (e.g. from mutagenic chemicals or radiation) or proof reading errors in the copying process. However, most people are unaware that there is significant DNA turnover in non-growing (non-dividing) cells (16). Adaptive (directed) mutation are mutations that apparently result in the selective appearance of favorable mutations. The designation of these mutations has caused considerable controversy and they have been called adaptive, directed, Cairnsian, selection-induced, and stressful lifestyle associated mutations (SLAM). One researcher coined the name "Fred" while trying to find a name that would not inflame the critics, and "Fred" seems to have found it's way into at least informal discourse by the relevant researchers (17). "Fred" occurs only under non-lethal selection in non-dividing cells, and has been suggested to be a neo-Lamarkian mechanism for getting environmental information into the genes. However, "Fred" is no such thing (16,17,18), and is not directed in the sense that Spetner suggests.
So the following model shows how "adaptive" mutations occur. Nutritionally deprived non-growing cells are under stress, stress leads to double stranded breaks in the DNA, recombination vis RecBCD primes the DNA synthesis, DNA PolIII finishes the job but makes mistakes, which slip through because starvation has largely turned off mismatch repair. This results in genome wide mutation at a faster rate than normal, and the occasional mutant that can utilize the "selective" substrate (16, 17, 18). The canonical example is E. coli that have a crippled Lac gene, and cannot utilize lactose, when plated on a medium that has only lactose as a carbon source, the cells cannot grow, but after a time colonies appear that can use lactose. These colonies contain a version of the crippled gene that has been restored to function (usually by a 1 bp frameshift (16, 17, 18). So Fred, while certainly exciting from the genetic point of view, turns out just another boring random mutation. Conclusion:So to summarize, although Spetner's arguments are superficially plausible, a deeper look with some knowledge of biochemistry shows massive flaws. Spetner is wrong in the details of the biology, ligand specificity is not directly governed by binding string length as required by Spetner's theory, and ligand binding is not an "all or nothing affair". This invalidates his analyses. Even then, Spetner's own examples do not support his claims. Furthermore, when using his metrics Spetner swaps metrics when one shows inconvenient changes. References:1. Spetner, L. M. (1998) NOT BY CHANCE! Shattering the Modern Theory of Evolution, Judaica Press, New York. 2. Shannon CE (1948) A Mathematical Theory of Communication. Bell Sys Techl J 27. 3. Schneider TD, (2000) Evolution of Biological Information, Nucleic Acids Res, 28(14): 2794-2799. 4. Max E (2001) The Evolution of Improved Fitness 5. Hunyady et al., (2003) Agonist induction and conformational selection during activation of a G-protein-coupled receptor. [ PubMed] TIPS 24, 81-86 6. Le MT et al., (2002) Angiotensin IV Is a Potent Agonist for Constitutive Active Human AT1 Receptors. J Biol Chem, 277, 23107-23110 7. Knowle D, et al., (2001) Role of Asp297 of the AT2 receptor in high-affinity binding to different peptide ligands. [ PubMed] Peptides, 22, 2145-2149 8. Turner, CA, et al., (1999) Role of the His273 Located in the Sixth Transmembrane Domain of the Angiotensin II Receptor Subtype AT2 in Ligand Receptor Interaction. [ PubMed] BBRC, 257, 704-707 9. Hartley, B.S. (1984) Experimental evolution of ribitol dehydrogenase. In R.P. Mortlock (ed.), "Microorganisms as Model Systems for Studying Evolution" (pp.23 - 54) Plenum, New York. 10. Matsumura I, Ellington AD. In vitro evolution of beta-glucuronidase into a beta-galactosidase proceeds through non-specific intermediates. [ PubMed] J Mol Biol. 2001 Jan 12;305(2):331-9 11. Hayashi Y, et al., Can an arbitrary sequence evolve towards acquiring a biological function? [ PubMed] J Mol Evol. 2003 Feb;56(2):162-8. 12. Björkman J, et al., Novel ribosomal mutations affecting translational accuracy, antibiotic resistance and virulence of Salmonella typhimurium. Mol Microbiol. 1999 Jan;31(1):53-8. 13. Yockey HP. (1992) Information Theory and Molecular Biology, Cambridge University Press. Chapter 6.3 14. Tolvonen JM et al., Mol. Micro. 1999, 1735-1746 15. Björkman J et al., (1998) Virulence of antibiotic resistant Salmonella typhimurium. Proc Natl Acad Sci USA. 95, 3949-3953. 16. Foster PL & Rosche WA. Mechanisms of mutation in nondividing cells. Insights from the study of adaptive mutation in Escherichia coli [ PubMed] Ann N Y Acad Sci 1999, 870, 133-45. 17. Rosenberg, SM. Mutation for Survival [ PubMed] Curr Opinion Gen Dev 1997, 7:829-834 18. Rosche WA, and Foster PL. (1999 Jun 8). The role of transient hypermutators in adaptive mutation in Escherichia coli. Proc Natl Acad Sci U S A , 96, 6862-7. Acknowledgments:Many thanks to Chris Ho-Stuart, Michael Hopkins, Bill Hudson and Douglas Theobald for helpful suggestions and proof reading. Originally posted at Talk Origins. |
